

Yetkili Kuruluş : Sağlık Bakanlığı
İlgili AB Direktifi : In vitro diagnostic medical devices
İlgili Yönetmelik : 98/79/EC Vücut Dışında Kullanılan Tıbbi Tanı Cihazları Yönetmeliği
İletişim: 0212 250 99 09 – info@cebelgesi.com
98/79/EC Vücut Dışında Kullanılan Tıbbi Tanı Cihazları Yönetmeliği (In Vitro Diagnostic Medical Devices Directive – IVD), Avrupa Birliği (AB) tarafından oluşturulmuş, vücut dışında teşhis amacıyla kullanılan tıbbi cihazların güvenli ve etkili bir şekilde kullanılmasını sağlamak için geliştirilen bir yönetmeliktir. Yönetmelik, AB içinde bu cihazların pazara sunulması, dağıtılması ve kullanımını düzenlemekte, cihazların kalite ve güvenlik standartlarına uygun olduğundan emin olunmasını sağlamaktadır.
Vücut Dışında Kullanılan Tıbbi Tanı Cihazı Nedir?
Vücut dışında kullanılan tıbbi tanı cihazları, insan vücudundan alınan örnekler üzerinde yapılan testlerle hastalıkların teşhis edilmesine yardımcı olan cihazlardır. Bu cihazlar laboratuvar testlerinden evde yapılan testlere kadar geniş bir yelpazeye yayılır. Kan, idrar, tükürük gibi biyolojik örnekler üzerinde testler yapan cihazlar, tanı koyma sürecinde hayati bir rol oynar. Örneğin, hamilelik testleri, kan şekeri ölçüm cihazları ve laboratuvarlarda kullanılan çeşitli tanı testleri bu kapsama girer.
98/79/EC Yönetmeliğinin Amacı
Bu yönetmeliğin temel amacı, in vitro tanı cihazlarının güvenliğini ve etkinliğini sağlamaktır. Yönetmelik, cihazların AB pazarında serbest dolaşımını sağlarken, aynı zamanda hastalar ve kullanıcılar için yüksek düzeyde güvenlik ve koruma sunar. Cihazların doğru sonuçlar vermesi ve bu sonuçların insan sağlığına katkı sağlaması büyük önem taşır.
98/79/EC Yönetmeliğinin Kapsamı
98/79/EC yönetmeliği, aşağıdaki ürünleri kapsar:
- In Vitro Tanı Cihazları: İnsan vücudundan alınan örnekler üzerinde analiz yaparak hastalıkları teşhis eden cihazlar. Örneğin, kan testleri, genetik analizler ve enfeksiyon testleri.
- Aksesuarlar: Tanı cihazlarıyla birlikte kullanılan, cihazın performansını doğrudan etkileyen tüm aksesuarlar da bu yönetmelik kapsamına girer. Bu aksesuarlar, test sırasında gerekli olan tüpler, test şeritleri gibi malzemeleri içerebilir.
- Kendi Kendine Test Yapılan Cihazlar: Evde yapılan hamilelik testi ve kan şekeri ölçüm cihazları gibi, bireylerin sağlık durumlarını kendi kendilerine izleyebilmelerini sağlayan cihazlar da bu yönetmelikte yer alır.
Cihaz Sınıflandırması
98/79/EC yönetmeliği, cihazları risk seviyelerine göre sınıflandırır. Bu sınıflandırma, cihazların kullanım amacına, kullanım yerine ve olası sonuçlarının insan sağlığı üzerindeki etkisine göre yapılır. Üç ana sınıf vardır:
- Genel Tanı Cihazları: Düşük riskli cihazlar olarak kabul edilir. Bu cihazlar, genel biyolojik analizlerde kullanılan testleri içerir.
- Kendi Kendine Test Cihazları: Evde kullanım için tasarlanmış, kişilerin kendi sağlık durumlarını izleyebildikleri cihazlardır. Hamilelik testleri ve kan şekeri ölçüm cihazları bu sınıfa girer.
- Liste B ve Liste A Cihazları: Liste B cihazları, bazı daha karmaşık testleri ve hastalıkları teşhis etmek için kullanılır. Liste A cihazları ise HIV ve hepatit gibi bulaşıcı hastalıkların teşhisinde kullanılan yüksek riskli cihazlardır.
CE Belgesi ve 98/79/EC Yönetmeliği
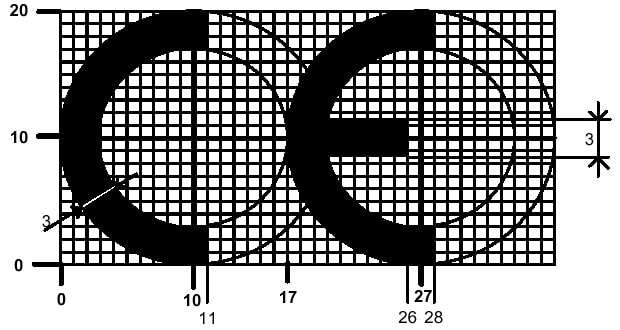
98/79/EC yönetmeliğine tabi olan tıbbi cihazların Avrupa pazarında serbest dolaşım yapabilmesi için CE işareti taşımaları zorunludur. CE işareti, cihazın yönetmelikte belirtilen güvenlik ve performans gerekliliklerine uygun olduğunu gösterir. Bir cihazın CE işareti taşıması, onun AB’nin belirlediği teknik ve güvenlik standartlarına uygun olduğu anlamına gelir.
CE belgesi almak için üreticinin aşağıdaki adımları takip etmesi gerekir:
- Risk Analizi: Cihazın kullanımı sırasında olası riskler belirlenir ve bu riskler en aza indirilir. Üretici, cihazın güvenli olduğuna dair gerekli kanıtları sunmalıdır.
- Uygunluk Değerlendirmesi: Cihazın AB standartlarına ve yönetmeliğin gerekliliklerine uygun olup olmadığını belirlemek için gerekli testler ve incelemeler yapılır.
- Teknik Dosya: Cihazın tasarımı, üretim süreci, test sonuçları ve güvenlik değerlendirmelerini içeren bir teknik dosya hazırlanır.
- Uygunluk Beyanı: Üretici, cihazın ilgili yönetmeliğe uygun olduğunu ve tüm gereklilikleri karşıladığını beyan eden bir uygunluk beyannamesi hazırlar.
- Onaylanmış Kuruluş Değerlendirmesi (Gerekli Durumlarda): Yüksek risk taşıyan cihazlar için bağımsız bir onaylanmış kuruluş tarafından ek değerlendirmeler yapılması gerekebilir.
98/79/EC Yönetmeliğine Uygun Olmayan Cihazlar İçin Yaptırımlar
Eğer bir cihaz 98/79/EC yönetmeliğine uygun değilse veya CE işareti taşımıyorsa, bu cihazların AB pazarında satışına izin verilmez. Uygunsuz cihazların pazardan çekilmesi, para cezaları ve üreticinin hukuki sorumlulukları gibi yaptırımlar uygulanabilir. Cihazların yanlış teşhise veya yanlış tedaviye yol açması durumunda, ciddi sağlık sorunlarına ve yasal sonuçlara yol açabilir.
98/79/EC Yönetmeliğinin Yerini Alan Yeni Yönetmelik
98/79/EC yönetmeliği, 26 Mayıs 2022 itibarıyla 2017/746/EU In Vitro Tıbbi Tanı Cihazları Yönetmeliği ile yer değiştirmiştir. Yeni yönetmelik, daha katı güvenlik ve kalite gereksinimlerini içermekte ve risk yönetimi, klinik değerlendirme ve piyasa gözetimi süreçlerini güçlendirmektedir. Bu yeni yönetmelik, tıbbi tanı cihazları endüstrisinde daha şeffaf ve güvenli bir ortam yaratmayı hedeflemektedir.
98/79/EC Vücut Dışında Kullanılan Tıbbi Tanı Cihazları Yönetmeliği, in vitro tanı cihazlarının güvenli ve etkili olmasını sağlayan bir düzenlemedir. Yönetmelik, bu cihazların kullanıcılar ve hastalar üzerinde risk oluşturmasını engellerken, AB pazarında serbest dolaşımını sağlar. CE işareti taşıyan cihazlar, bu güvenlik gerekliliklerini karşılayarak tıbbi tanı sürecinde önemli bir rol oynar. 2022 itibarıyla yerini alan yeni yönetmelik, bu cihazların güvenliğini daha da artırarak sağlık sektöründe daha güvenilir bir altyapı oluşturmayı amaçlamaktadır.






{kind=link}